
Both DNA and RNA viruses are dependent of cell
proliferation for replication. Thus viruses often posses proteins which are
able to interact with cellular proteins which are responsible for maintaining
the regulation of the cell cycle. In general viruses could either slow the cell
cycle or activate the cell cycle.
 |
| Figure: Viruses and the DDR: multiple points of action |
In the case of HIV-1, the viral R protein (VpR ) was
identified to be responsible for accumulation of G2 arrested cells. by preventing the
activation of the cyclinB/CDK 1 through
the inhibition of a phosphatase, cdc25A, and the activation of a kinase, Wee1. Both
enzymes are hypophosphorylated in HIV-1 infected cells and VpR may act through
the activation of protein phosphatase 2A (PP2A)
and thus blocks the G2-M
transition. In this context it is very interesting to note that VpR -similar to
coronaviral N protein is also located in the nucleolus of the cell. Another
example is the paramyxovirus Simian parainfluenza virus 5, which is able to
slow the progression both G1 into S and G2 into M but not of a G1 or G2 arrest
.The V protein of Simian Virus 5 binds to a protein involved in DNA repair,
DDB1 (DNA damage binding protein). DDB1 becomes activated upon binding through
V. Activation of DDB1 in turn leads to
slowing the progression of G2 into M and binds to the Retinoblastoma protein
and thus prevents entering of the S phase through non-activation of E2F.
Because of the delayed activation of E2F, the transactivation of the genes
necessary for successful entry into the S phase is also delayed.
Coronavirus and the cell cycle: Induction of p53 by
ATR induces S phase arrest
The expression of the nucleocapsid (N) protein of
Infectious Bronchitis Virus (IBV) or Severe Acute Respiratory Syndrome Virus
(SARS) in Vero cells decreases not only cell proliferation in the absence of
apoptosis but also arrests the cell cycle at S phase as well as inducing in a
significant number of cells a mitotic defect as indicated by the presence of a
cleavage furrow.
 |
| Figure: Localisation of Coronavirus N protein and mitotic arrest |
Indeed, bivariate flow
cytometry of BrdU incorporation and Propidium iodide content and subsequent dot
plot analysis by gating for G0/G1, S and G2/M phase showed that the number of
cells in S phase increased by 20% if the cells were transfected with IBV N
compared to mock transfected or cells transfected with empty vector as well as
an increase in cells with DNA content higher than 4n,indicating not only that
the expression of IBV N delays the cell cycle but also might induce mitotic
defects. Similar the cell cycle is delayed in Vero cells arrested in G2/M with
Nocadazole and released for 24 hrs by replacing the Nocadazole containing
medium with fresh DMEM, indicating that the expression of N in Vero cells does
delay the progression of Vero cells from S phase into G2 phase of the cell
cycle. In Vero cells infected with a Vero adapted IBV Beaudette US strain a
similar delay can also be observed (albeit to a lower extent) indicating that
viral replication does not antagonise the ability of N to delay the cell cycle.
In contrast to Vero cells, the expression of IBV N in Saos-2 cells that are
deficient for p53 does not induce a delay or arrest in the cell cycle
indicating that p53 is required for inducing the observed delay in cell cycle
progression.
 |
| Figure: Expression of IBV N in Vero cells but not in Saos-2 cells induces cell cycle arrest that it can be abrogated by Caffeine |
p53 can be activated by ATR as a result of nucleolar
stress and since the expression of the expression of the coronaviral N protein
has been proposed to induce nucleolar stress, treatment of cells transfected
with SARS or IBV N with an inhibitor of ATR should therefore reverse the
activation of p53 by ATR. Indeed, Vero cells transfected with either IBV or SARS
N protein and treated with 4 mM Caffeine do not exhibit an
increase in S phase -indicating that N
protein induced S phase arrest is indeed mediated by an activation of ATR- but
instead exhibit an increase in G0/G1 phase of the cell cycle indicating that N
inhibits the progression of cells from G1 into S phase. Indeed, the expression
of N does not only induce nucleolar stress but also inhibits Cyclin D1 as well
as Cyclin A and E thus inhibiting entry into S phase in cells treated with
Caffeine.
 |
| Figure: The expression of SARS N in A549 cells induces a transient arrest in S phase |
In addition, Caffeine treatment of Vero cells
transfected with IBV N also increases the percentage of cells in G2/M
indicating the activation of the G2/M checkpoint and flow cytometry analysis of
Caffeine treated Vero cells transfected with SARS N confirms that the
expression of SARS N induces a G2 rather than a mitotic arrest since SARS N
does not increase Histone H3(Ser-10) phosphorylation.
In conclusion, the expression of IBV and SARS N
induces an arrest primary in S phase of the cell cycle that is at least partially
dependent on the induction of p53 by ATR. Abrogation of ATR dependent cell
cycle arrest induces an arrest in late G1/early S phase and an arrest in G2
-but not M- phase.
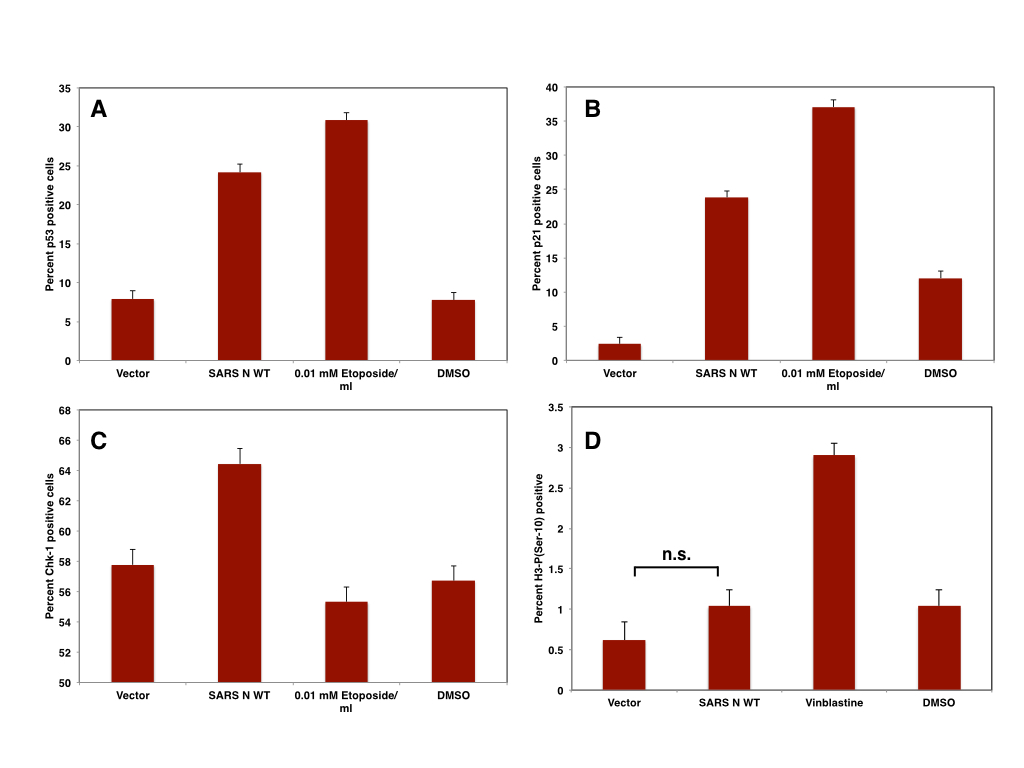 |
| Figure: The expression of SARS N in Vero cells induces p53 (A), p21 (B), CHK-1 (C) and but not mitotic arrest (D) |
As discussed before, the expression of N might induce
DRAM-1 and thus autophagy as a result of nucleolar stress that might promote
viral replication by promoting the formation of viral replication centres. In budding yeast, ATR mediated induction of
autophagy has been linked to the induction of anaphase and recent evidence suggests
that LC3 positive structures -probably mitophagosomes- are present throughout
mitosis. Induction of autophagy by N therefore might be responsible for a
premature onset of anaphase and thus inducing the formation of the midbody.
Since aberrant cytokinesis is only present in a subset of cells expressing N,
the majority of cells might not undergo premature mitosis but instead, the
induction of autophagy might be a contributing factor to N induced S-G2/M
arrest since autophagy can arrest cells by increasing levels of phosphorylated
Cdc2(Y15), Cyclin A, E and B1, and Rb (S807/811) and decreasing Cyclin D1.
Furthermore, Coronavirus N induced autophagy might also attenuate the DNA
damage response by degrading Chk-1 via chaperone-mediated autophagy.
In addition to N, the expression of IBV and SARS
nsp-13 has been shown to induce the DNA damage response as well as inducing S
phase arrest via ATR. In this case, ATR is activated by stalled replication
forks due to the inhibition of p125
subunit of DNA polymerase δ and -similar to IBV and SARS N- the activation of
p53 might induce autophagy. Since nsp-13 localises both to the nucleus and the
cytoplasm, nsp-13 might trigger distinct events at different times of the viral
replication cycle. ER localised nsp-13 might trigger the ER stress response and
akin to (and maybe in addition to) nsp-4 and -6, promote the formation of viral
RC by initiating the formation of omegasome-like structures. Nuclear nsp-13 in
contrast inhibits DNA replication and induces the ATR response which in turn
might facilitate the formation of RC indirectly by inducing the formation of
autophagosomes and/or by inducing a S-G2/M arrest thus preventing degradation
of the viral genome during mitosis as well as extending the cell cycle to allow
viral replication. The individual contributions of both N and nsp-13 are
however not known and it remains to be seen if the localisation of nsp-13 is
altered during viral replication, i.e. if nsp-13 undergoes nuclear-cytoplasmic
shuttling. It should also be noted that the expression of SARS N induces the
activation of caspase-3 in the absence of depolarised mitochondria and in the
absence of apoptosis. N and/or nsp-13 induced autophagy therefore might also
promote the degradation of proapoptotic proteins or promote mitophagy and thus
prevent Coronavirus induced apoptosis in addition to promoting the formation of
viral RC. Interestingly, activated Caspase-3 localises to the nucleolus in A549
cells transfected with SARS N without -as indicated by staining of SARS N
transfected cells with HSP-60 and Bcl-2 as well as flow cytometry detection of
activated Bax- the presence of damaged mitochondria.
 |
| Figure: SARS N induces the localisation of active Caspase-3 to the nucleolus |
 |
| Figure: Activity of Caspase-3 is not impaired in A549 cells transfected with SARS N |
 |
| Figure: SARS N does not impair mitochondrial integrity in A549 cells |
In human MRC-7 cells the disintegration of the
nucleolus has been shown to precede cell cycle arrest and p53 activation, suggesting
that the nucleolar localisation of active Caspase-3 might contribute to
nucleolar stress induced by the expression of SARS-CoV N protein, but further
studies are needed to determine the pathway of caspase-3 activation and if
damaged mitochondria are degraded by mitophagy. Also, it has to be determined
of the induction of ATR by N or any other coronaviral protein does prevent the
activation of caspase-3 dependent apoptosis. Alternatively, the expression of
nsp-13 or N might induce the ER stress response and thus protects cells from
caspase-3 dependent apoptosis.
In conclusion, both the expression of N and nsp-13
induces the ATR dependent DNA damage response as well might prevent the
execution of the DDR by abrogating downstream signaling pathways thus arresting
cells in S and/or G2 phase of the cell cycle. In the case of N, inhibition of
G1 and S phase cyclins, namely Cyclin D1, A, and E, contributes to the observed
arrest. The induction of p53 via ATR by N or nsp-13 might however contribute to
the induction of Interferon type I signaling, which can be counteracted by the
expression of the PLP2 catalytic domain
of nsp-3 . PLP2 therefore might not only inhibit antiviral singling but also
partially prevent N induced cell cycle arrest, namely pathways induced by p53
such as the degradation of CHK-1 and the increase of Cyclin E m A, and B as
well as the decrease of Cyclin D1, thus preventing S phase arrest in addition
to prevent mitotic defects. So far however this has not been experimentally
been proven.
 |
| Figure: IBV N, nsp-13, and nsp-14: induction of ATR and ATM dependent pathways |
Furthermore, it needs to be determined why the
expression of the TGEV N protein in contrast to the N protein derived from IBV,
SARS, or MHV does induce p53 dependent apoptosis. Similar to SARS N, TGEV N
induces a cell cycle arrest (in S and G2/M) and the activation of Caspase-3;
unlike in A549 cells expressing SARS N however, Bax translocates to the
mitochondria and induces the depolarisation of the mitochondrial membrane. It
might be possible that in PK-15 autophagy is impaired and damaged mitochondria
might accumulate. One possibility is that both SARS and IBV N do increase the
expression of both Bak and Mcl-1 whereas TGEV N does not.
Finally, it needs to be determined if the nucleolar
localisation of other viral proteins such as the core protein of JEV induces
the activation of ATR as a result of nucleolar stress.

Further reading
Flynn RL, & Zou L (2011). ATR: a master conductor of cellular responses to DNA replication stress. Trends in biochemical sciences, 36 (3), 133-40 PMID: Myers K, Gagou ME, Zuazua-Villar P, Rodriguez R, & Meuth M (2009). ATR and Chk1 suppress a caspase-3-dependent apoptotic response following DNA replication stress. PLoS genetics, 5 (1) PMID: 19119425
Rawlinson SM, & Moseley GW (2015). The nucleolar interface of RNA viruses. Cellular microbiology, 17 (8), 1108-20 PMID: 26041433
Wulan WN, Heydet D, Walker EJ, Gahan ME, & Ghildyal R (2015). Nucleocytoplasmic transport of nucleocapsid proteins of enveloped RNA viruses. Frontiers in microbiology, 6 PMID: 26082769
Xu LH, Huang M, Fang SG, & Liu DX (2011). Coronavirus infection induces DNA replication stress partly through interaction of its nonstructural protein 13 with the p125 subunit of DNA polymerase δ. The Journal of biological chemistry, 286 (45), 39546-59 PMID: 21918226
Wang, F., Fei, H., Cui, Y., Sun, Y., Li, Z., Wang, X., Yang, X., Zhang, J., & Sun, B. (2014). The checkpoint 1 kinase inhibitor LY2603618 induces cell cycle arrest, DNA damage response and autophagy in cancer cells Apoptosis, 19 (9), 1389-1398 DOI: 10.1007/s10495-014-1010-3
Dove B, Brooks G, Bicknell K, Wurm T, & Hiscox JA (2006). Cell cycle perturbations induced by infection with the coronavirus infectious bronchitis virus and their effect on virus replication. Journal of virology, 80 (8), 4147-56 PMID: 16571830
Liou JS, Wu YC, Yen WY, Tang YS, Kakadiya RB, Su TL, & Yih LH (2014). Inhibition of autophagy enhances DNA damage-induced apoptosis by disrupting CHK1-dependent S phase arrest. Toxicology and applied pharmacology, 278 (3), 249-58 PMID: 24823293
Wrighton, K. (2015). Autophagy: Chaperone-mediated autophagy degrades CHK1 Nature Reviews Molecular Cell Biology, 16 (6), 328-328 DOI: 10.1038/nrm4003
Filippi-Chiela EC, Villodre ES, Zamin LL, & Lenz G (2011). Autophagy interplay with apoptosis and cell cycle regulation in the growth inhibiting effect of resveratrol in glioma cells. PloS one, 6 (6) PMID: 21695150
Lee IH, Kawai Y, Fergusson MM, Rovira II, Bishop AJ, Motoyama N, Cao L, & Finkel T (2012). Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science (New York, N.Y.), 336 (6078), 225-8 PMID: 22499945
Wojciechowski J, Horky M, Gueorguieva M, & Wesierska-Gadek J (2003). Rapid onset of nucleolar disintegration preceding cell cycle arrest in roscovitine-induced apoptosis of human MCF-7 breast cancer cells. International journal of cancer. Journal international du cancer, 106 (4), 486-95 PMID: 12845642
Xu L, Khadijah S, Fang S, Wang L, Tay FP, & Liu DX (2010). The cellular RNA helicase DDX1 interacts with coronavirus nonstructural protein 14 and enhances viral replication. Journal of virology, 84 (17), 8571-83 PMID: 20573827
Han C, Liu Y, Wan G, Choi HJ, Zhao L, Ivan C, He X, Sood AK, Zhang X, & Lu X (2014). The RNA-binding protein DDX1 promotes primary microRNA maturation and inhibits ovarian tumor progression. Cell reports, 8 (5), 1447-60 PMID: 25176654
Li FQ, Tam JP, & Liu DX (2007). Cell cycle arrest and apoptosis induced by the coronavirus infectious bronchitis virus in the absence of p53. Virology, 365 (2), 435-45 PMID: 17493653
Yuan L, Chen Z, Song S, Wang S, Tian C, Xing G, Chen X, Xiao ZX, He F, & Zhang L (2015). p53 degradation by a coronavirus papain-like protease suppresses type I interferon signaling. The Journal of biological chemistry, 290 (5), 3172-82 PMID: 25505178
Ding L, Huang Y, Du Q, Dong F, Zhao X, Zhang W, Xu X, & Tong D (2014). TGEV nucleocapsid protein induces cell cycle arrest and apoptosis through activation of p53 signaling. Biochemical and biophysical research communications, 445 (2), 497-503 PMID: 24548406
Huang Y, Ding L, Li Z, Dai M, Zhao X, Li W, Du Q, Xu X, & Tong D (2013). Transmissible gastroenteritis virus infection induces cell apoptosis via activation of p53 signalling. The Journal of general virology, 94 (Pt 8), 1807-17 PMID: 23677787
No comments:
Post a Comment