Zika Virus (ZIKV) was first isolated from monkeys in 1947 and until 2007 only isolated cases of infection in humans were reported although serologic studies suggested widespread distribution in Africa and Southeast Asia. In 2007 however ZIKV of the Asian lineage caused an outbreak in Yap/Federal Micronesia followed by an outbreak in French Polynesia in 2013 and the current outbreak in Central & South America and the Caribbean. ZIKV is mainly transmitted via infected Aedes spp.mosquitoes but sexual transmission via semen of infected man has also been reported in addition to contaminated blood and blood products.
Clinical symptoms caused by ZIKV infection are very similar to that of other arboviral infections such as Dengue Virus (DENV) or Chikungunya Virus (CHIKV) which often co-circulate in areas affected by ZIKV and more often than not ZIKV infection is asymptomatic. In general, ZIKV may cause fever, maculopapular rash, and arthralgia or conjunctivitis. Following the outbreak in French Polynesia 2013 -2014 an increase in Guillain-Barré Syndrome (GBS) has been observed (albeit only by retrospective analysis) and -based on data obtained during the current outbreak-a link between the onset of microcephaly and ZIKV infection during the early stages of gestation has been proposed (but not verified).
Experimentally, the neurotrophic properties of ZIKV have been demonstrated in mice injected with mouse adapted strain of ZIKV intraperitoneally and a isolates directly of obtained from monkeys that were injected intracelebral. In both cases, virus replicated in neuronal cells of the CNS and the brain with extensive apoptosis of infected cells. A subtraction of infected mice also showed paralysis or morbidity, suggesting that in mice ZIKV can induce a severe infection. Cerebrospinal fluid from GBS patients has been shown to induce neuronal apoptosis suggesting the presence of neurotic factors, but at present this has not been confirmed to be the case for ZIKV induced GBS and the relevance to disease pathology is unclear. In the case of ZIKV induced microcephaly, ZIKV may be transmitted to the embryo and/or foetus and interfering with brain development via the placenta. Indeed, ZIKV has been isolated in the placenta of ZIKV positive pregnant women, suggesting that ZIKV might be transmitted via exosomes (similiar to HCV) that infect embryonic and/or foetal cells of the neuroepithelium or directly infects the cerebral cortex during the earliest stages of brain development. Alternatively, ZIKV infection of the placenta disrupts the outer layer of the placenta thus inducing either miscarriage or -in the absence of viral induced miscarriage- contribute to viral induced microcephaly by disrupting placental signals to the developing brain and inducing an inflammatory response with the developing foetus (similar to MHV-68). In any case, ZIKV has been detected both in the amniotic fluid of at least two pregnant women whose foetuses were diagnosed with microcephaly as well as in foetal brain tissue, suggesting that the currently circulating ZIKV strains are indeed neurotrophic and might contribute to the onset of microcephaly.
Experimentally, younger mice infected with ZIKV derived from monkey exhibit more severe brain damage compared to older mice suggesting that ZIKV indeed might indeed interfere with the developing brain. Human induced pluripotent stem cells differentiated into forebrain specific human neural progenitor cells (hNPC) that have been infected with the original ZIKV strain MR766 used in the mice studies published in 1952 , suggest that ZIKV induces cell cycle arrest and apoptosis (and subsequent growth arrest) 72 hrs p.i. .
Gene expression analysis of ZIKV infected hNPC showed that ZIKV infection alters the expression of at least 7000 genes related to cell cycle regulation, the DNA damage response, autophagy, apoptosis and other cellular processes suggesting that ZIKV might indeed interfere with these pathways as discussed before and as detailed below. In general, ZIKV infection downregulates the expression of genes encoding for proteins regulating DNA repair and cell cycle progression whilst upregulating the expression of genes ending for proteins regulating autophagy and the ER stress response, suggesting that ZIKV induces the formation of stalled replication forks, inhibiting the progression of S to G2 phase, inducing mitotic arrest whilst promoting the formation of lipid droplets and autophagosome-like vesicles thus supporting viral replication and dissemination.
ZIKV: inhibition of DDR, DNA replication and the cell cycle
As discussed before, both retroviruses and positive strand RNA viruses (in addition to DNA viruses interfere with the DNA damage response which can be induced by the expression of viral proteins, endogenous reactive oxygen species or by exogenous agents including UV and chemotherapeutic drugs. Depending on the nature of the DNA damage, different repair pathways can be induced, with the homologous DNA repair pathway (HR) predominant in S and G2 cells and the Non-Homologous End Joining (NHEJ; which itself can be distinguished between a conservative and alternative pathway) present in all phases of the cell cycle, (probably) including M phase. Upon successful DNA repair, cells reenter the cell cycle whereas incomplete DNA repair can induce an arrest in S phase and subsequent G2 arrest; failure to activate the G2/M checkpoint may result in mitotic arrest followed by apoptosis or abberant mitosis, thus contributing to the development of tumour cells.
 |
| Figure: General outline of the connection between the DNA damage response, cell cycle regulation and autophagy |
In the case of positive RNA viruses, the expression of IBV and SARS nsp13 protein has been shown to induce stalled replication forks and the expression of the N protein derived from different Coronaviruses has been shown to delay the progression from S to G2 probably by inducing ATR (and thus potentially the ATR dependent DNA damage response). Delaying the onset of G2 might favour viral replication not only by extending G2 and thus providing enough time to replicate but also provide cellular resources such as enzymes or nucleotides required for viral replication. Failure to resolve DNA damage that naturally occurs during DNA replication or persistent activation of the DDR by viral proteins however might contribute also to mitotic arrest and thus induce apoptosis.
 |
| Table: Genes that are down-or unregulated in ZIKV infected cells: DDR |
In the case of ZIKV infected hNPC, viral infection decreases the expression of genes encoding proteins that are required for at least three distinct pathways, namely HR dependent pathways induced by both ATM and ATR, as well as the POLQ dependent Base-Excision Repair (BER) pathway and the Fanconi Anemia (FA) pathway that intersects with the ATR pathway. In contrast to the decreasing the expression of genes related to the main DNA damage repair pathways, the expression of at least one gene, STKL18, related to the Nucleotide Excision Repair and a NBS1 independent repair pathway, ATMIN, is increased following ZIKV infection. The relative contribution of these pathways to DNA repair in general are however minor and if these pathways are functional in ZIKV infected cells is not known presently.

 |
| Figure: ZIKV potentially inhibits the DDR at multiple sites whilst inducing the acetylation of NBS1 and the initial recruitment of sensors to sites of DNA damage induced by stalled DNA replication |
Similar to genes encoding proteins regulating the DDR, the expression of genes encoding proteins regulating the cell cycle, in particular genes related to the progression of G1, G1 to S, S to G2 and those related to the control of mitotic progression and chromosome segregation, are generally downregulated with the notable exception of NEDD1. Since NEDD1 encodes for a protein involved in the duplication of the centriole and spindle assembly, it might be possible that the over expression of NEDD-1 induced by ZIKV infection might induce mitotic arrest due to the presence of multiple centrioles. In addition to genes encoding for proteins such as Cyclin B1, CDK2, or CDC25A, ZIKV expression also downregulates the expression of genes related to DNA replication, suggesting that ZIKV infection might also induce the formation of stalled replication forks, which could be measured experimentally using a CldU/IdU based assay.
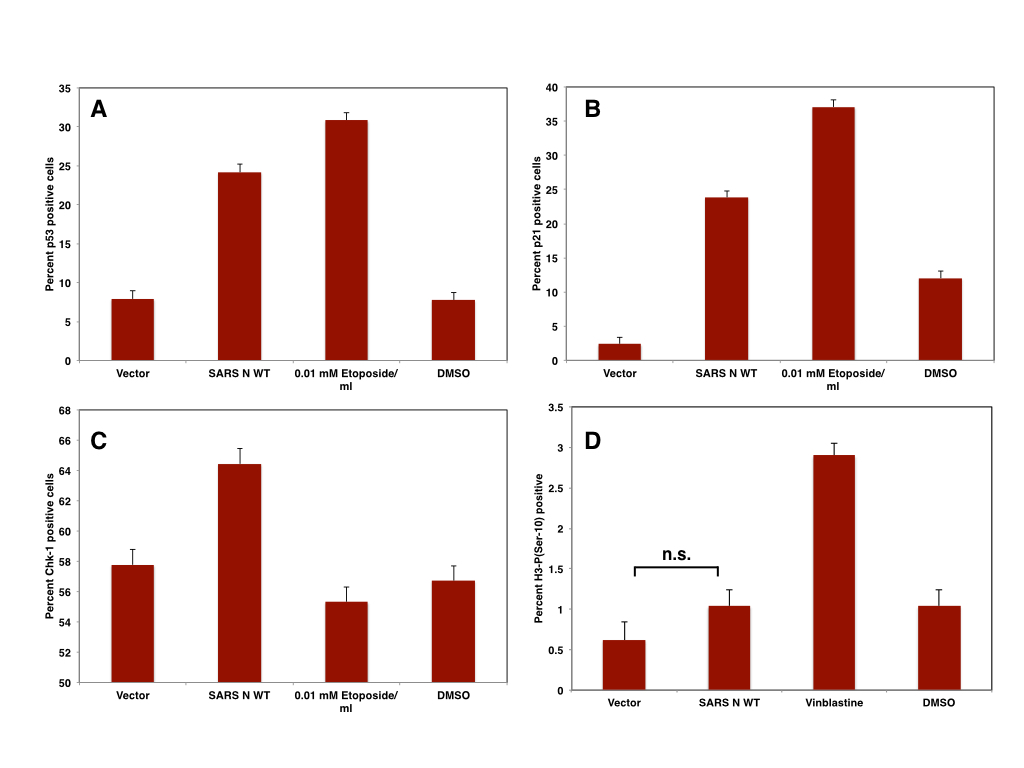
Consequently, it can be assumed that ZIKV infection induces the arrest of infected cells in G2 and/or M phase of the cell cycle -which is supported by flow cytometry based cell cycle analysis of infected hNPC cells. Using Histone H3-P(Ser-10) as a marker for mitotic cells it should be easy to discriminate cells arrested in G2 from cells arrested in M phase. Confocal microscopy can be used to further distinguish cells arrested in prophase from those in metaphase or anaphase. Based on the data obtained from analysing the transcriptome, it might be however possible that ZIKV arrests cells in multiple stages of mitosis, since not only the formation of spindle poles might be affected but also the progression of anaphase and thus the onset of telophase.
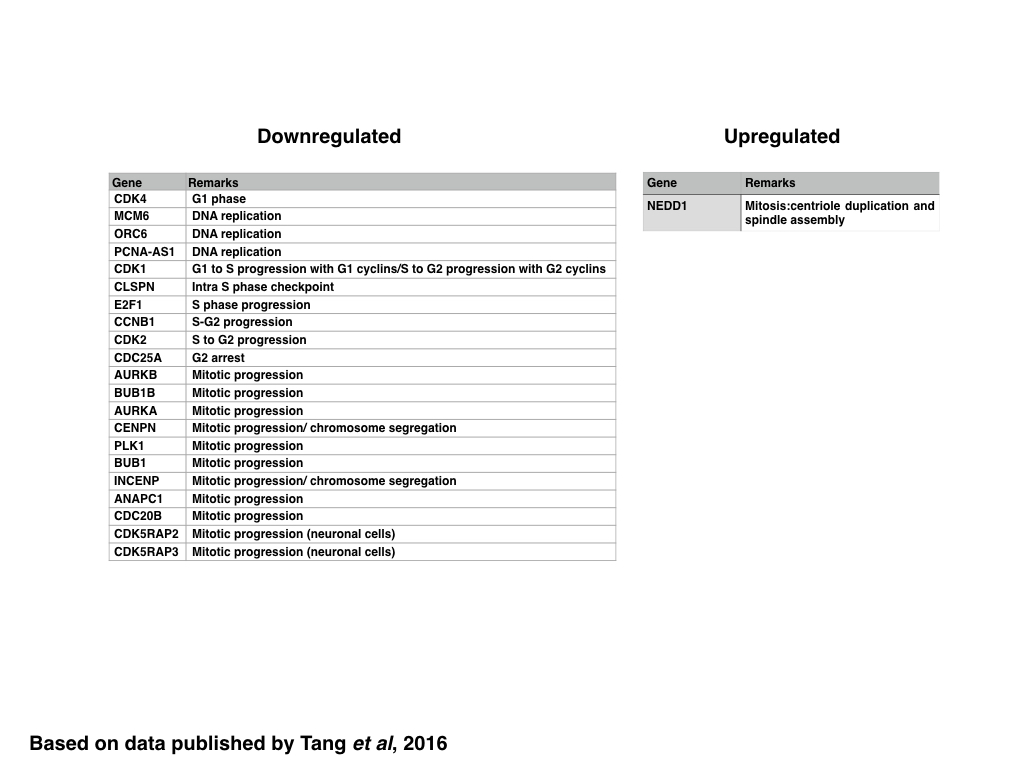 |
| Table: Genes down- or upregulated in ZIKV infected cells: cell cycle |
 |
| Figure: Cell Cycle arrest in ZIKV infected cells |
ZIKV: promotion of autophagosome formation, ER stress response and lipid droplet formation
In contrast to genes related to the control of the cell cycle, the DDR and DNA replication, ZIKV infection generally induces the expression of genes encoding for proteins required for the initiation of autophagosome formation including those encoding for proteins that increase the pool of cytosolic LC3 (SIRT1 and ATG4A) or induce the formation of autophagosomes (STK38L, ATG16L1, RABGAP1 and ULK1) whereas selective (p62/SQSTM-1 dependent) autophagy is inhibited maybe due to decreasing the expression of KLF4 and stress induced autophagy by decreasing the expression of Caspase-2. The maturation of the autophagosome and/or lysosome however might be inhibited by decreasing the expression of LAMP2, similar to the formation of antiviral autophagic clusters by decreasing the expression of ISG15.
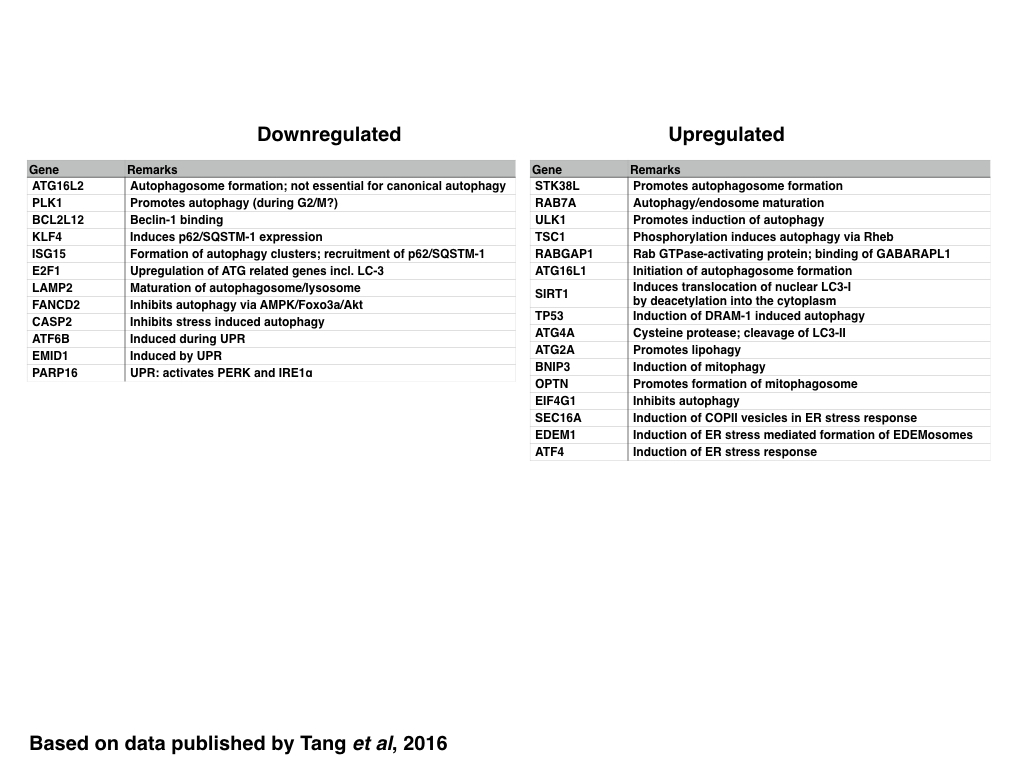 |
| Table: Genes are down-orupregualted in ZIKV infected cells: Autophagy and UPR |
 |
| Figure: ZIKV inhibits selective autophagy whilst promoting the formation of viral RC via increased autophagosome/EDEMosome formation |
Increasing the expression of Optineurin and BNIP-3 might promote the formation of mitophagsomes and thus lipophagy as described before. The synthesis of lipid droplets at the ER might be aided by increased expression of DCP2 and XRN1, and similar to cells infected with HCV, ZIKV might promote the formation of ER localised lipid droplets by decreasing the number of P-bodies whilst decreasing the formation of stress granules.
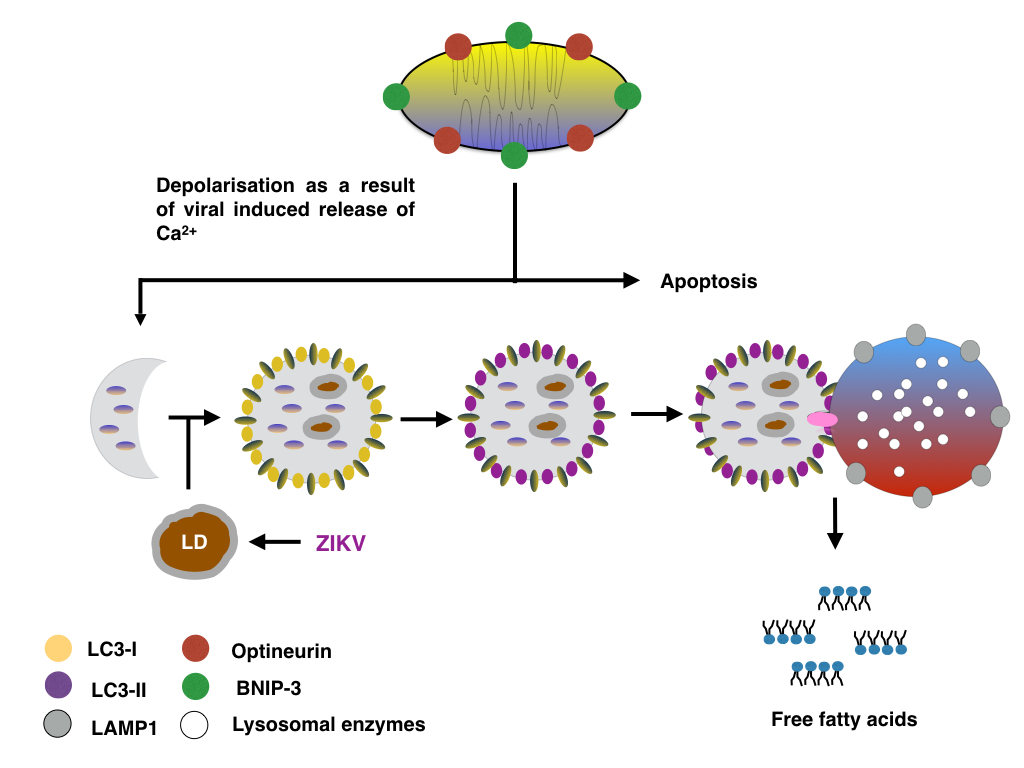 |
| Figure: Lipid droplets are degraded via ZIKV induced mitophagy |
The formation of lipid droplets and autophagosomes in ZIKV infected cells might also be supported by increasing the expression of proteins regulating the ER stress response/Unfolded Protein Response (UPR) such as SEC16A (inducing the formation of COPII vesicles) and EDEM1 (inducing EDEMosomes). In the case of BNIP-3, increased BNIP-3 might either induce mitophagy or induce the release of Ca2+ from the ER and thus promote mitochondrial apoptosis; alternatively BNIP-3 might induce the depolarisation whilst the Optineurin facilitates the clearance of damaged mitochondria by mitophagy.
The induction go both the PERK and IRE-1 induced ER stress however might be inhibited, thus preventing -at least partially- the induction of UPR induced apoptosis. In contrast, only the ATF mediated ER stress response might be still active, resulting in delayed or decreased expression of CHOP.
 |
| Figure: ZIKV and the ER stress response |
In summary, ZIKV might induce the formation of autophagosomes or autophagosome-like structures by inducing the expression of proteins that are known inducers of autophagosome formation as well as inducing at least a partial ER stress response whilst inhibit selective autophagy. The formation of autophagosomes and the inhibition of p62/SQSTM-1 dependent selective autophagy has been confirmed in skin fibroblasts and keratinocytes whilst the induction of the UPR by ZIKV has not been demonstrated yet. In addition to inducing autophagosomes, the formation of antiviral clusters might be inhibited,similar to neuronal cells infected with Yellow Fever Virus, probably due to the downregulation of both ISG15 and p62/SQSTM-1 expression. Propagation of lipid droplet formation and lipophagy by decreasing the number of P bodies and promoting the formation of mitophagosomes fused with LD's might promote viral replication by providing free fatty acids but probably not by providing a scaffold for the viral replication complex (in contrast to HCV). If the upregulation of SEC16A contributes to the release of exsomes containing viral particles similar to HCV remains also to be seen.
 |
| Figure: ZIKV induced pathways are linked, controlling cell proliferation in multiple ways |
In conclusion, ZIKV induces apoptosis by regulating by generally downregulating the expression of genes encoding proteins that regulate the replication of cellular DNA, the DNA damage response and the cell cycle as well as upregulating genes expressing proteins that are proapoptotic (DIABLO and FADD). Apoptosis is therefore induced by an arrest in G2 and M phase of the cell cycle as well as promoting the depolarisation of mitochondria. Inducing the expression of genes that promote autophagosome formation as well as formation and degradation of lipid droplets in contrast promotes viral replication.The spatial expression pattern of these genes however remains to be elucidated, but in general the analysis of the transcriptome on ZIKV infected hNPC cells provides an insight in the complex interaction of the various pathways regulated by ZIKV and ZIKV related viruses.
Further reading
DICK GW, KITCHEN SF, & HADDOW AJ (1952). Zika virus. I. Isolations and serological specificity. Transactions of the Royal Society of Tropical Medicine and Hygiene, 46 (5), 509-20 PMID: 12995440
DICK GW (1952). Zika virus. II. Pathogenicity and physical properties. Transactions of the Royal Society of Tropical Medicine and Hygiene, 46 (5), 521-34 PMID: 12995441
Calvet, G., Aguiar, R., Melo, A., Sampaio, S., de Filippis, I., Fabri, A., Araujo, E., de Sequeira, P., de Mendonça, M., de Oliveira, L., Tschoeke, D., Schrago, C., Thompson, F., Brasil, P., dos Santos, F., Nogueira, R., Tanuri, A., & de Filippis, A. (2016). Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: a case study The Lancet Infectious Diseases DOI: 10.1016/S1473-3099(16)00095-5
Brasil, P., Pereira, Jr., J., Raja Gabaglia, C., Damasceno, L., Wakimoto, M., Ribeiro Nogueira, R., Carvalho de Sequeira, P., Machado Siqueira, A., Abreu de Carvalho, L., Cotrim da Cunha, D., Calvet, G., Neves, E., Moreira, M., Rodrigues Baião, A., Nassar de Carvalho, P., Janzen, C., Valderramos, S., Cherry, J., Bispo de Filippis, A., & Nielsen-Saines, K. (2016). Zika Virus Infection in Pregnant Women in Rio de Janeiro — Preliminary Report New England Journal of Medicine DOI: 10.1056/NEJMoa1602412
Chan JF, Choi GK, Yip CC, Cheng VC, & Yuen KY (2016). Zika fever and congenital Zika syndrome: An unexpected emerging arboviral disease? The Journal of infection PMID: 26940504
Adibi, J., Marques, E., Cartus, A., & Beigi, R. (2016). Teratogenic effects of the Zika virus and the role of the placenta The Lancet DOI: 10.1016/S0140-6736(16)00650-4
Bell TM, Field EJ, & Narang HK (1971). Zika virus infection of the central nervous system of mice. Archiv fur die gesamte Virusforschung, 35 (2), 183-93 PMID: 5002906
Tang, H., Hammack, C., Ogden, S., Wen, Z., Qian, X., Li, Y., Yao, B., Shin, J., Zhang, F., Lee, E., Christian, K., Didier, R., Jin, P., Song, H., & Ming, G. (2016). Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth Cell Stem Cell DOI: 10.1016/j.stem.2016.02.016
Biegel JM, & Pager CT (2016). Hepatitis C Virus Exploitation of Processing Bodies. Journal of virology PMID: 26937026
Katzenell S, & Leib DA (2016). Herpes simplex virus and interferon signaling induce novel autophagic clusters in sensory neurons. Journal of virology PMID: 26912623
Ryan EL, Hollingworth R, & Grand RJ (2016). Activation of the DNA Damage Response by RNA Viruses. Biomolecules, 6 (1) PMID: 26751489
Riz I, Hawley TS, & Hawley RG (2015). KLF4-SQSTM1/p62-associated prosurvival autophagy contributes to carfilzomib resistance in multiple myeloma models. Oncotarget, 6 (17), 14814-31 PMID: 26109433
Park JM, Choi JY, Yi JM, Chung JW, Leem SH, Koh SS, & Kang TH (2015). NDR1 modulates the UV-induced DNA-damage checkpoint and nucleotide excision repair. Biochemical and biophysical research communications, 461 (3), 543-8 PMID: 25912875
J
Joffre C, Codogno P, Fanto M, Hergovich A, & Camonis J (2016). STK38 at the crossroad between autophagy and apoptosis. Autophagy, 1-2 PMID: 26890257
Joffre C, Dupont N, Hoa L, Gomez V, Pardo R, Gonçalves-Pimentel C, Achard P, Bettoun A, Meunier B, Bauvy C, Cascone I, Codogno P, Fanto M, Hergovich A, & Camonis J (2015). The Pro-apoptotic STK38 Kinase Is a New Beclin1 Partner Positively Regulating Autophagy. Current biology : CB, 25 (19), 2479-92 PMID: 26387716
Kanu N, & Behrens A (2007). ATMIN defines an NBS1-independent pathway of ATM signalling. The EMBO journal, 26 (12), 2933-41 PMID: 17525732
Matsui A, Kamada Y, & Matsuura A (2013). The role of autophagy in genome stability through suppression of abnormal mitosis under starvation. PLoS genetics, 9 (1) PMID: 23382696
Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, Perera-Lecoin M, Surasombatpattana P, Talignani L, Thomas F, Cao-Lormeau VM, Choumet V, Briant L, Desprès P, Amara A, Yssel H, & Missé D (2015). Biology of Zika Virus Infection in Human Skin Cells. Journal of virology, 89 (17), 8880-96 PMID: 26085147
Beller M, Thiel K, Thul PJ, & Jäckle H (2010). Lipid droplets: a dynamic organelle moves into focus. FEBS letters, 584 (11), 2176-82 PMID: 20303960
Bukong TN, Momen-Heravi F, Kodys K, Bala S, & Szabo G (2014). Exosomes from hepatitis C infected patients transmit HCV infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PLoS pathogens, 10 (10) PMID: 25275643
Shrivastava S, Devhare P, Sujijantarat N, Steele R, Kwon YC, Ray R, & Ray RB (2015). Knockdown of Autophagy Inhibits Infectious Hepatitis C Virus Release by the Exosomal Pathway. Journal of virology, 90 (3), 1387-96 PMID: 26581990
Schorey JS, Cheng Y, Singh PP, & Smith VL (2015). Exosomes and other extracellular vesicles in host-pathogen interactions. EMBO reports, 16 (1), 24-43 PMID: 25488940